
Zespoły mielodysplastyczne
Ta grupa chorób nowotworowych cechuje się zaburzeniem funkcjonowania szpiku kostnego. To bardzo niejednorodna grupa: niektóre choroby przebiegają gwałtownie i nieleczone szybko prowadzą do śmierci chorego, inne mają przebieg przewlekły.
Hanna Mądra
Konsultacja: prof. dr hab. n. med. Joanna Góra-Tybor, Kierownik Oddziału Chorób Układu Krwiotwórczego Kliniki Hematologii, Instytut Hematologii i Transfuzjologii w Warszawie
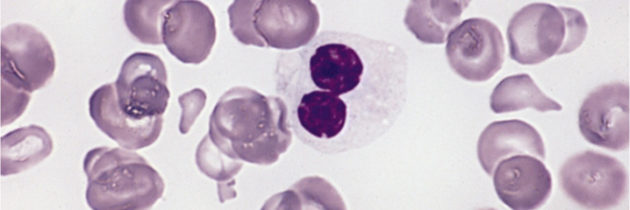
Na zespoły mielodysplastyczne (MDS) zapada co roku więcej osób, niż na białaczki. Mimo to są one mniej znane lekarzom. To sprawia, że są wykrywane rzadko i często za późno. Jest to grupa chorób, w których dochodzi do zmniejszenia liczby krwinek czerwonych, białych i/lub płytek krwi we krwi obwodowej wskutek zaburzeń procesu krwiotworzenia w szpiku kostnym. Wszystkie te komórki powstają w szpiku kostnym z obecnych w nim pierwotnych komórek macierzystych. W MDS dochodzi do uszkodzenia tych pierwotnych komórek szpiku, wskutek czego komórki krwi, które powstają i dojrzewają, są nieprawidłowe. Oglądane pod mikroskopem wykazują nieprawidłowe cechy budowy, czyli są dysplastyczne. Stąd bierze się nazwa choroby.
Etiologia
Na MDS zapadają przede wszystkim osoby starsze: aż 80 proc. przypadków zachorowań diagnozuje się w grupie osób po 60. roku życia (częściej u mężczyzn). W ogólnej populacji na MDS choruje pięć osób na każde 100 tys. rocznie, a wśród osób starszych jest to już 22-45 na każde 100 tys. W Polsce z chorobami wchodzącymi w zakres MDS walczy kilka tysięcy osób. W zespołach mielodysplastycznych dochodzi do uszkodzenia materiału genetycznego komórek macierzystych w szpiku. Wiemy już, że do takiego uszkodzenia może dojść pod wpływem kontaktu z substancjami toksycznymi takimi jak benzen, pestycydy, herbicydy czy rozpuszczalniki. Ważne dla palaczy papierosów: niektóre z substancji, które są podejrzewane o wywoływanie zespołów mielodysplastycznych, występują w dymie papierosowym! Ryzyko zachorowania na MDS zwiększa także promieniowanie jonizujące i leki cytostatyczne, stosowane w terapii nowotworów. Zdarza się, że wiele lat po leczeniu raka u chorego może się rozwinąć jako odległe powikłanie właśnie jakiś rodzaj zespołu mielodysplastycznego. Bardzo często jednak nie udaje się ustalić czynnika, wywołującego chorobę. Choroby te mają skłonność do tzw. progresji, czyli – jeśli się już rozwiną – nie wycofają się same i z czasem będą się nasilać. Tempo tej progresji bywa powolne, ale bywa też i gwałtowne. Jednak głównym problemem w MDS jest to, że istnieje ryzyko transformacji tej jednostki chorobowej w ostrą białaczkę szpikową.
Objawy i rozpoznanie
Rozpoznanie MDS utrudnia fakt, że objawy choroby są mało charakterystyczne, początki są zwykle zupełnie bezobjawowe. Kiedy objawy wystąpią, prawie wszyscy chorzy skarżą się na osłabienie, zmęczenie, zawroty głowy i problemy z koncentracją. Mają gorszą tolerancję wysiłku, nękają ich trudne do wyleczenia zakażenia grzybicze i bakteryjne. Niemalże połowa cierpi z powodu skazy krwotocznej: po niewielkich urazach (a nawet bez urazów) pojawiają się sińce, krwawienia oraz wybroczyny na skórze i śluzówkach.
Najczęściej podejrzenie MDS pada na podstawie obrazu morfologii krwi obwodowej. Badanie to ujawnia z reguły małopłytkowość, leukopenię z neutropenią czy niedokrwistość przebiegającą z prawidłowym lub powiększonym rozmiarem krwinki czerwonej. Zawsze należy wówczas wykluczyć takie przyczyny zmian w morfologii jak niedobór żelaza, kwasu foliowego czy witaminy B12.
Przy braku jasnej przyczyny zmian we krwi konieczna jest pogłębiona diagnostyka obejmująca biopsję aspiracyjną szpiku kostnego (materiał do badania pobiera się zwykle z talerza kości biodrowej, rzadziej z mostka) oraz trepanobiopsję szpiku kostnego. Niezbędna jest ocena nieprawidłowości w procesie dojrzewania komórek hematopoetycznych, oszacowanie odsetka blastów w szpiku, ocena komórkowości, a także obecność (lub brak) pierścieniowatych syderoblastów i włóknienia.
Leczenie
Całkowite wyleczenie MDS możliwe jest wyłącznie poprzez przeszczepienie szpiku od dawcy rodzinnego lub niespokrewnionego. Zanim jednak zapadnie decyzja o takiej metodzie leczenia, trzeba rozważnie przeanalizować wszystkie za i przeciw. Przede wszystkim należy ocenić rokowanie w danym rodzaju zespołu MDS, wziąć pod uwagę wiek pacjenta i choroby współistniejące. Trzeba pamiętać, że przeszczepienie szpiku kostnego to metoda obarczona dużym ryzykiem powikłań, u 20-30% chorych będących przyczyną śmierci. Stąd wciąż trwa poszukiwanie metod leczenia zachowawczego, które umożliwi przedłużenie życia starszych pacjentów, poprawę jego jakości oraz kontrolę parametrów morfologii krwi (złagodzenie niedokrwistości) i infekcji towarzyszących. Jedną z metod leczenia jest przetaczanie koncentratów krwinek czerwonych (KKCz), inną – podawanie erytropoetyny czy przetaczanie koncentratu płytek krwi. W razie neutropenii i ciężkich zakażeń podaje się czynnik wzrostu granulocytów. Jednak i takie leczenie jest uciążliwe i niebezpieczne, ponieważ przetaczanie krwi trzeba wykonywać (u niektórych chorych) nawet co 2 tygodnie. To może prowadzić do wtórnego przeładowania żelazem, grożącego uszkodzeniem serca, stawów, wątroby czy trzustki (i związaną z tym cukrzycą).
Na szczęście, w ostatnim dziesięcioleciu widać wyraźny postęp w leczeniu MDS. Postęp ten zawdzięczamy głównie lepszemu i precyzyjniejszemu diagnozowaniu (choć wielu chorych wciąż dowiaduje się o swej chorobie przez przypadek, podczas rutynowych badań kontrolnych) oraz coraz nowocześniejszym metodom leczenia. U chorych z zaawansowanymi postaciami MDS, z dużą liczbą blastów leczeniem przynoszącym przejściową poprawę jest azacytydyna.
W rzadkich przypadkach MDS z obecnością charakterystycznej zmiany chromosomalnej – izolowanej delecji 5q bardzo skutecznym lekiem jest lenalidomid. Zespół 5q minus cechuje się niewielką obecnością blastów w szpiku kostnym (poniżej 5 proc.) oraz często nadpłytkowością. Lenalidomid został zatwierdzony do leczenia chorych z MDS przez Amerykańską Agencję ds. Żywności i Leków (FDA, Food and Drug Administration) już w 2005 roku. Chorzy, by zakwalifikować się do leczenia lenalidomidem muszą spełniać pewne warunki: mieć izolowaną delecję 5q minus, być zależni od przetoczeń krwinek czerwonych oraz zostać zakwalifikowani do grupy niskiego i średniego ryzyka według IPSS (International Prognostic Scoring System). Zastosowanie lenalidomidu pozwala na całkowite uniezależnienie się od przetoczeń u połowy pacjentów. Leczenie lenalidomidem to leczenie przewlekłe i zaleca się kontynuowanie terapii tak długo, jak długo jest ona skuteczna. Lenalidomid ma jednak działania niepożądane. Choć wiek pacjenta nie ma negatywnego wpływu na skuteczność samej terapii, ma jednak wpływ na częstość występowania objawów niepożądanych. Do takich skutków należą neutropenia i małopłytkowość wymagające nieraz czasowej przerwy w leczeniu czy zmniejszenia dawki leku. Jeśli parametry morfologiczne spadną poniżej pewnych norm (w przypadku granulocytów poniżej 500 × 106/l, w przypadku małopłytkowości poniżej 50 × 109/l) terapię należy przerwać – ale gdy parametry wrócą do normy, można wrócić do podawania lenalidomidu. W przypadku chorych z podwyższonym ryzykiem zakrzepowo-zatorowym należy równolegle z lenalidomidem stosować profilaktykę aspiryną lub drobnocząsteczkową heparyną.

 Instytut Nagrody Zaufania Złoty OTIS
Instytut Nagrody Zaufania Złoty OTIS